
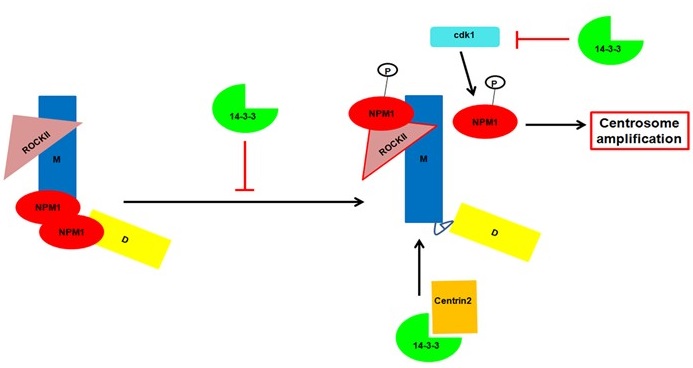
The 14-3-3 protein family in mammals is a conserved set of small acidic proteins that regulate several cellular pathways by forming a complex with ligands containing a phosphorylated serine residue in one of two consensus motifs. However, the generation of ligand specificity for the different 14-3-3 isoforms and the processes by which the 14-3-3 proteins bind to and dissociate from their ligands is still unclear. Our previous work had demonstrated that while all 14-3-3 proteins bind to their ligands in vitro, not all of them bind to specific ligands in vivo and we have demonstrated that this is due to the ability of the 14-3-3 proteins to discriminate between ligands in cells. Previous work from the laboratory had demonstrated that loss of either 14-3-3ε or 14-3-3γ in the colon cancer cell line HCT116 leads to centrosome amplification, which was associated with an increase in the activity of cdc25C and thus an increase in cdk1 activity. Further, we demonstrated that completely disrupting complex formation between cdc25C and 14-3-3 proteins led to tumor regression in xenograft assays in mice suggesting that this complex could serve as a potential therapeutic target. We have also demonstrated in proteome based screens that 14-3-3 proteins bind to multiple centrosomal proteins suggesting that 14-3-3 proteins might play multiple roles in centrosome biogenesis and duplication. 14-3-3 proteins are required for the localization of Centrin2 to the centrosomes, a crucial step in centriole biogenesis. We have recently identified two conserved acidic amino acid residues, an Aspartic acid (D129) and a Glutamic acid (E136) at residues 129 and 136 in 14-3-3γ, in the peptide-binding pocket of 14-3-3 proteins that contribute to ligand binding. Altering these residues to Alanine results in changes in function leading to defects in centriole duplication in multiple cell types as cells expressing the D129A mutant showed an increase in the number of mitotic cells with a single centrosome while cells expressing the E136A mutant showed an increase in the number of cells with >2 centrosomes. A double mutant showed a phenotype similar to WT 14-3-3γ suggesting that these mutants had opposing effects resulting in intragenic complementation. We demonstrated that the 14-3-3 proteins inhibit the function of Nucleophosmin 1 (NPM1), which inhibits centrosome duplication and phosphorylation of NPM1 at T199 by cdk1 or cdk2 is a licensing event for centrosome duplication. We have observed similar phenotypes upon expression of similar mutants in 14-3-3ε (D127A and E134A). However, the double mutant in 14-3-3ε showed a phenotype similar to the D127A mutant suggesting that the D127A mutation was dominant over the E134A mutation. Thus, in this highly conserved region of this protein family, the same mutants induce different cellular phenotypes suggesting that these phenotypic differences may be due to differential ligand binding or differences in the roles of these conserved amino acids in different 14-3-3 proteins. Most of these results are summarized in figure 2.
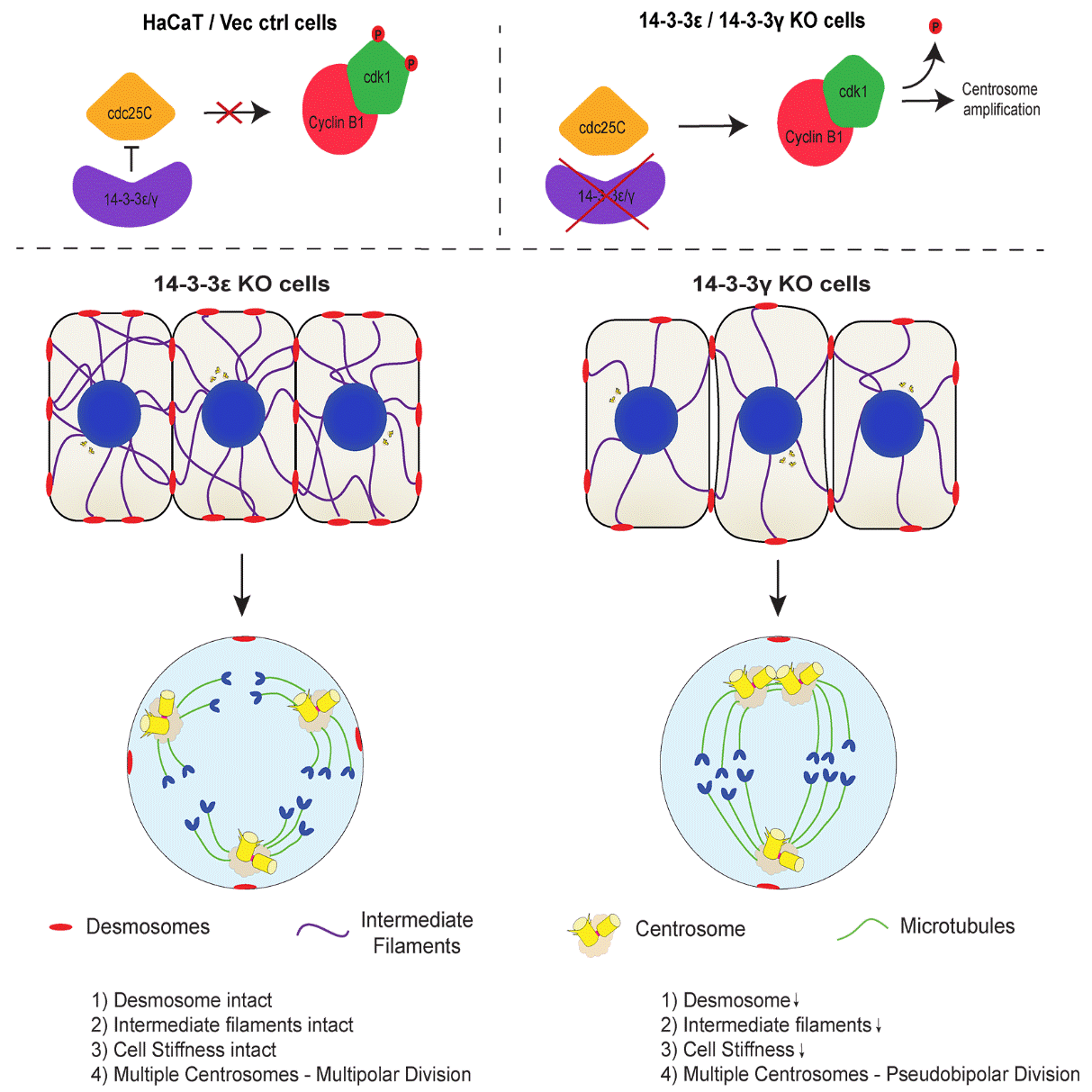
Loss of 14-3-3ε or 14-3-3γ leads to centrosome amplification in human cells. However, only the loss of 14-3-3γ leads to clustered mitoses while cells lacking 14-3-3ε undergo multipolar mitoses. The results in this report demonstrate that cells lacking 14-3-3γ exhibit compromised desmosome function leading to a decrease in the attachment of keratin filaments to desmosomes at the cell border and decreased cell stiffness as compared to cells lacking 14-3-3ε. Restoration of desmosome formation in the 14-3-3γ knockout cells by over-expressing a border localized plakoglobin construct, led to an increase in multi-polar mitoses in these cells. Similarly, a knockdown of either plakoglobin or keratin5 in the 14-3-3ε knockout cells leads to a decrease in desmosome function and cell stiffness and an increase in the frequency of cells undergoing clustered mitoses (figure 3). These results highlight a novel role of desmosome function in regulating centrosome separation and demonstrate that phenotypes observed upon 14-3-3 loss reflect the dis-regulation of multiple pathways.